




(Michal Parzuchowski, Unsplash)
23 Oct. 2023. We’re traveling for the next two weeks, and will resume our editorial posts on Monday 6 Nov. 2023.
* * *
|
|||||
|
|
(Michal Parzuchowski, Unsplash) 23 Oct. 2023. We’re traveling for the next two weeks, and will resume our editorial posts on Monday 6 Nov. 2023. * * * 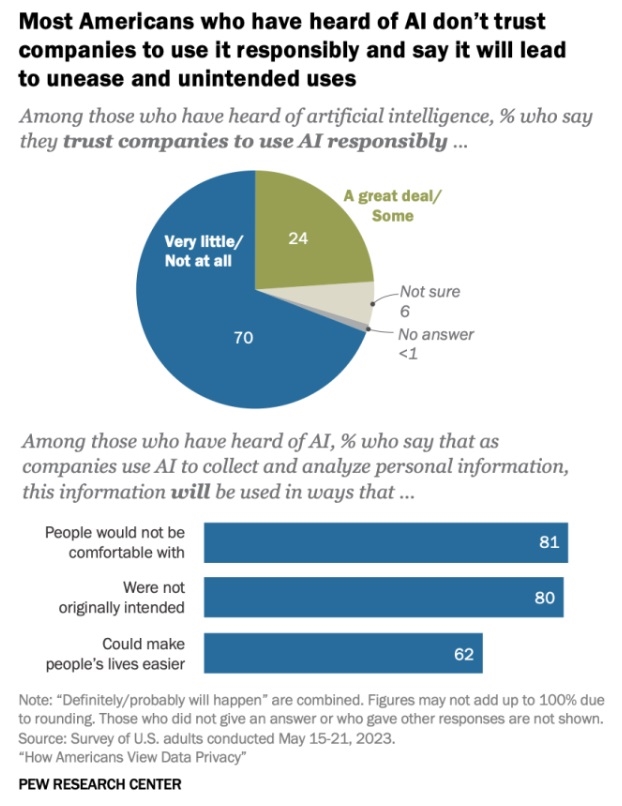 Click on image for full-size view. (Pew Research Center) 21 Oct. 2023. A poll in the U.S. shows a large majority of Americans who heard of artificial intelligence have very little or no trust that companies will use the technology responsibly. The Pew Research Center revealed the findings this week in a report on Americans’ views of A.I. and related issues of data privacy. The poll, taken among American adults in May 2023, shows seven in 10 (70%) in the U.S. who have heard of A.I. have very little or no trust at all that companies will use A.I. responsibly. Only about a quarter of respondents who heard of A.I. (24%) have a great deal or at least some trust in companies using A.I. in a responsible manner, with the remainder unsure or not answering. In separate questions, even larger majorities of those who heard of A.I. — about eight in 10 — believe companies will use A.I. in ways that were not originally intended (80%) or will make people uncomfortable (81%). Fewer, but still a majority (62%) say companies’ use of A.I. could make people’s lives easier. Pew Research Center conducted the survey online with 5,101 U.S. adults taking part in the Center’s American Trends Panel from 15 to 21 May 2023. Participants are recruited through national random sampling of U.S. households, with results weighted to be representative of the U.S. adult population. More from Science & Enterprise:
We designed Science & Enterprise for busy readers including investors, researchers, entrepreneurs, and students. Except for a narrow cookies and privacy strip for first-time visitors, we have no pop-ups blocking the entire page, nor distracting animated GIF graphics. If you want to subscribe for daily email alerts, you can do that here, or find the link in the upper left-hand corner of the desktop page. The site is free, with no paywall. But, of course, donations are gratefully accepted. * * *
 (Colin Behrens, Pixabay. https://pixabay.com/illustrations/hand-molecule-chemistry-science-898232/) 20 Oct. 2023. A life science venture investor is merging two of its biotechnology portfolio companies to form a single developer of nanoscale medications with programmable properties. Flagship Pioneering in Cambridge, Massachusetts is combining its portfolio companies Laronde and Senda Biosciences, both in Cambridge, Mass., to form a new business, Sail Biomedicines. Flagship Pioneering says the new company will combine the programmable elements of Laronde’s synthetic RNA therapeutics technology with Senda Bio’s process harnessing nanoscale particles from natural microorganisms to deliver treatments programmed to reach and execute inside cells. Laronde creates synthetic RNA therapeutics that overcome limitations of natural messenger RNA, the nucleic acid carrying instructions from the genetic code to cells to produce proteins. The company adapts RNA from non-coding regions of the genome to form circular RNA strands that it says are more stable and long-lasting than natural RNA. That circular RNA, called endless RNA by Laronde, is then modified and designed to express amino acids that form into peptides, antibodies, enzymes, or receptor proteins with specific properties. Science & Enterprise reported on Laronde’s emergence from stealth mode in May 2021, and raising $50 million in initial financing. Organisms co-evolved with human cellsSenda Biosciences also creates programmable therapeutics, but with a technology it says uses characteristics from four types of natural microorganisms in bacteria, archaea — single cell organisms lacking a nucleus but different from bacteria, fungi, and plant cells. Senda Bio says these nanoscale organisms co-evolved with human cells and thus have chemical properties that can be exploited for therapeutics programmed to reach specific cells, then execute designed actions within cells. That programming, says Senda Bio, is done with synthetic messenger RNA, delivering instructions for cells to generate specific proteins. Flagship Pioneering says Sail Biomedicines plans to combine the stability and long-acting properties of circular RNA from Laronde with the programmable nanoparticle chemistry of Senda Bio to develop new therapies with the ability to reach, deliver, and execute inside cells as programmed. In addition, says Flagship, the new company expects to take advantage of generative artificial intelligence to apply its process to a wide range of therapies. Executives from Flagship Pioneering will serve as Sail Biomedicines initial leaders. John Mendlein, executive partner at Flagship also serving as CEO of Laronde, is the initial executive chair at Sail Biomedicines. “Endless RNA,” says Mendlein in a Flagship statement, “has the potential to create an entirely new class of programmable medicines across therapeutic areas that we will now be able to deliver directly to cells and tissues via deployment molecules with unique properties to confer specificity and greater tolerability.” Guillaume Pfefer, CEO and partner at Flagship and also CEO at Senda Bio is the CEO at Sail Biomedicines as well. “Our deployment platform,” says Pfefer, “utilizes natural nanoparticles to shuttle biomolecules into human cells, with unique tropism, potency, and redosability.” More from Science & Enterprise:
We designed Science & Enterprise for busy readers including investors, researchers, entrepreneurs, and students. Except for a narrow cookies and privacy strip for first-time visitors, we have no pop-ups blocking the entire page, nor distracting animated GIF graphics. If you want to subscribe for daily email alerts, you can do that here, or find the link in the upper left-hand corner of the desktop page. The site is free, with no paywall. But, of course, donations are gratefully accepted. * * *
 (National Human Genome Research Institute, NIH) 19 Oct. 2023. An historically Black medical college and pharmaceutical industry group began a program to increase the numbers of people of African descent in genomic databases. The initiative brings together Meharry Medical College in Nashville with four drug makers that aim to make research for drug development more representative of diverse populations, and also help people of African ancestry make better health decisions with their genetic data. The companies AstraZeneca, Novo Nordisk, and Roche with the Regeneron Genetics Center are joining with Meharry Medical College in the Together for CHANGE initiative — CHANGE stands for Changing Healthcare for People of African Ancestry through an InterNational Genomics & Equity. The project is managed by a newly formed group, Diaspora Human Genomics Institute or DHGI, cofounded by Meharry as its academic partner. The project seeks to address the low representation of people from Africa and descendants worldwide in genomic research, which leads to documented disparities in health outcomes. DHGI cites data showing less than two percent of genetic data today comes from people of African descent, leading to only a 25 percent rate of predicting disease risks in Black people based on genomic data. In addition, within African and descendants’ populations are a wide diversity of genetics, cellular, and molecular activity affecting risks of disease. The Together for CHANGE goal is to build a database of de-identified genomics data from up to 500,000 volunteer participants of African ancestry. And the project plans to link the data to electronic health records, as well as make the databases available to researchers at historically Black colleges and universities or HBCUs in the U.S. DHGI also recognizes the need for increased counseling for individuals offering their genomic data, by establishing a training program for counselors and a fellowship in medical genetics at HBCUs. An ethics committee of Black community leaders, says DHGI, will advise the project. First reference genome of African ancestryMoreover, Together for CHANGE says it’s supporting increased genetic representation by a public education campaign among people with African ancestry to make more informed decisions about their health and protect their interests when participating in medical research. Plus, the group plans to increase exposure of school-age students to science, technology, and health careers. Meharry Medical College formed the Together for CHANGE initiative with Regeneron Genetics Center, a division of the biopharmaceutical company Regneneron in Tarrytown, N.Y., as well as drug makers AstraZeneca, Novo Nordisk, and Roche. Each of the companies is making contributions valued at $20 million, with Regeneron Genetics Center or RGC also conducting genetic sequencing of submitted samples. “People of African ancestry have been underrepresented in genomics studies,” says Aris Baras, who heads the RGC in a company statement, “which leads to clinical genetic testing that has less reference data and less confident testing results.” Baras adds, “genetic databases function best as global resources when they reflect humanity’s broad spectrum of ethnic and genetic diversity, so that the resulting research and medical innovation may benefit all populations.” The project is also expected to spark more genomics research in Africa and at HBCUs. Anil Shanker, senior V.P. for research and innovation at Meharry Medical College notes that “researchers from historically Black colleges and universities and Africa will have access to the first-ever reference genome of African ancestry people to build collaborative projects at the intersection of genomics and health equity research that will ensure that the breakthroughs represent a healthier future for everyone, including the global Black communities that have historically been ignored.” More from Science & Enterprise:
We designed Science & Enterprise for busy readers including investors, researchers, entrepreneurs, and students. Except for a narrow cookies and privacy strip for first-time visitors, we have no pop-ups blocking the entire page, nor distracting animated GIF graphics. If you want to subscribe for daily email alerts, you can do that here, or find the link in the upper left-hand corner of the desktop page. The site is free, with no paywall. But, of course, donations are gratefully accepted. * * *
 Organ on a chip (Wyss Institute, Harvard Univ.) 18 Oct. 2023. The U.S. government’s health preparedness agency awarded a contract with a university lab to explore organ models on chips to document effects of radiation sickness. The Biomedical Advanced Research and Development Authority or BARDA awarded the contract to the Wyss Institute, a biomedical engineering research center at Harvard University, valued at $15.2 million to support the agency’s medical countermeasures against radiological and nuclear threats. BARDA, an agency of the U.S. Department of Health and Human Services funds diagnostics, vaccines, drugs, and other therapies to protect public health and prepare for strategic threats from chemical, biological, radiological, and nuclear weapons. Among the work supported by BARDA is preclinical studies of illnesses or injuries from these weapons, such as radiation sickness, known formally as acute radiation syndrome. Radiation sickness can occur after a large, direct, and penetrating nuclear radiation exposure, as experienced by survivors of the atomic bomb explosions in Japan during World War II and first responders to the Chernobyl nuclear power plant fire in 1986. This exposure penetrates into the organs, depleting functional stem cells in tissue, and affecting bone marrow, gastrointestinal organs, and the central nervous and cardiovascular systems, with multiple life-threatening symptoms. Legislation allows for animal testing alternativesThe Wyss Institute in Boston is a pioneer in developing lab-on-a-chip devices that simulate human organ functions. Language in the recently passed and signed FDA Modernization Act 2.0 allows for alternatives to animal testing in preclinical studies, such as microfluidic organ chips, small clear plastic devices with channels and wells lined with human cells. These chip devices, about the size of computer thumb drives, are designed to model and record actions of organ tissue under various conditions, bypassing scientific and ethical shortcomings of exposing animals to diseases or toxic experiences such as radiation exposure. Under the BARDA contract, Wyss Institute researchers plans to simulate radiation exposure that can cause acute radiation syndrome on several organ chips, including models of human bone marrow, lungs, lymph nodes, and intestines, where effects on the gut microbiome will be studied. The team is expected to analyze organ responses at the molecular level with changes to DNA, RNA, and proteins, as well as effects on cells, tissue, and immune functions. Donald Ingber, founding director of the Wyss Institute and lead investigator on the project, says in a statement that the BARDA award continues work for the Food and Drug Administration showing their organ chips can simulate human organs when exposed to radiation. “With this new expanded BARDA support,” says Ingber, “we will be extending this work to get insight into how radiation influences immune responses in the lymph node, and it will allow us to leverage these unique models in combination with A.I.-based computational discovery tools we have developed here at the Wyss, to discover new and more effective radiation countermeasure drugs” Inber is also the scientific founder and advisor to Emulate Inc., a company in Boston advancing research by the Wyss Institute on organ chips. Science & Enterprise has reported several times on Emulate Inc., most recently in Oct. 2020 on the FDA’s use of organ chip devices from Emulate Inc. to test mechanisms for preventing Covid-19 infections, as well as other processes. More from Science & Enterprise:
We designed Science & Enterprise for busy readers including investors, researchers, entrepreneurs, and students. Except for a narrow cookies and privacy strip for first-time visitors, we have no pop-ups blocking the entire page, nor distracting animated GIF graphics. If you want to subscribe for daily email alerts, you can do that here, or find the link in the upper left-hand corner of the desktop page. The site is free, with no paywall. But, of course, donations are gratefully accepted. * * *
 (LJNovaScotia, Pixabay) 17 Oct. 2023. A new company is underway that says it adapts the gene editing technique Crispr to make detecting nucleic acid targets like DNA or RNA faster and simpler. VedaBio in San Diego emerged from stealth mode with a process based on research in biomedical engineering at University of Illinois, and is raising $40 million in its first venture round. VedaBio is a two year-old business developing a process for molecular detection, such as identifying a suspected pathogen in a blood or saliva sample. The so-called gold standard for detecting molecular targets is polymerase chain reaction or PCR that takes a small sample of DNA and makes a large volume of copies to amplify the DNA sample for detailed sequencing and analysis. Using PCR today, however, usually requires expensive lab equipment and often a long turnaround time for analysis. The VedaBio technology it calls Crispr Cascade seeks to make that detection process faster and easier. The company’s process uses elements of the gene editing technique Crispr, short for clustered regularly interspaced short palindromic repeats, to guide gene-editing enzymes to specific locations and cleave the DNA. In this case, VedaBio uses a combination of RNA and protein as a guide to a precise DNA location, where a second RNA-protein combination makes a series of cuts in the DNA target to create a large volume of copies to amplify that target. For that amplifying to occur, the initial cuts in the target DNA generate a cascade effect to produce a high volume of cleavages, with those DNA pieces in the sample bound to reporter molecules for visual or electronic detection. Results in under one minuteAnurup Ganguli and colleagues discuss the technology in two patent applications filed in Dec. 2022 and Feb. 2023. The patent documents describe a process that returns detection results in less than a minute and in ambient temperatures of 16 degrees C (61 F) or less, for applications in research, medical diagnostics, environmental monitoring, and food safety. Ganguli is a co-founder and CEO of VedaBio, and previously served as a postdoctoral researcher and staff scientist in the biomedical engineering lab of Rashid Bashir at University of Illinois at Urbana-Champaign. Bashir, also a co-founder of VedaBio, and lab associates study micro- and nanotechnology applications in diagnostics and therapeutics, including microfluidics or lab-on-a-chip devices and biosensors for diagnostics. In July 2017, Science & Enterprise reported on a microfluidics device developed in the Bashir lab to quickly detect sepsis that the researchers tested with hospital patients. “With the Crispr Cascade reaction,” says Ganguli in a VedaBio statement, “we have unlocked the true power of Crispr with a platform that delivers near-instant molecular detection of highly multiplexed analytes with best-in-class accuracy, all without the need for target amplification.” VedaBio is raising $40 million in its first venture round, led by life science investor OMX Ventures in Lincolnshire, Illinois that says it invests mainly in early stage start-ups. Joining the round are venture investor Kleinmuntz Associates in Chicago and a group of undisclosed private investment managers known as family offices. According to PitchBook, VedaBio raised $1.15 million in seed funds in Aug. 2021. More from Science & Enterprise:
We designed Science & Enterprise for busy readers including investors, researchers, entrepreneurs, and students. Except for a narrow cookies and privacy strip for first-time visitors, we have no pop-ups blocking the entire page, nor distracting animated GIF graphics. If you want to subscribe for daily email alerts, you can do that here, or find the link in the upper left-hand corner of the desktop page. The site is free, with no paywall. But, of course, donations are gratefully accepted. * * *
 (Steve Sewell, Pixabay. https://pixabay.com/photos/superbike-motorsport-fast-speed-930715/) 16 Oct. 2023. A developer of an auto-injection technology says its device can deliver a biologic drug under the skin in a fraction of the time of conventional syringes. Presenters from Halozyme Therapeutics Inc. in San Diego are scheduled to deliver findings from a clinical trial tomorrow (17 Oct. 2023) at the Partnership Opportunities in Drug Delivery or PODD conference in Boston. Halozyme Therapeutics is a developer of alternative drug delivery technologies including subcutaneous devices for delivering drugs under the skin’s outer layers. The company’s main technology, called Enhanze, uses a synthetic enzyme that degrades hyaluronic acid, also known as hyaluronan, found in abundance in the extracellular matrix in skin and other soft connective tissues. Under the skin, says Halozyme, hyaluronan binds with water and forms a gel-like substance. And because of its relatively high volume, this gel acts as a barrier that physically slows the flow of liquids, including biologics and small molecule drug compounds. The Enhanze enzyme, says the company, is similar to natural hyaluronidases or hyaluronan-degrading enzymes, but is designed to encourage dispersion and absorption of drugs under the skin. A related synthetic enzyme is designed for cancer treatments to break down hyaluronan accumulations in the tumor microenvironment. Once under the skin, the enzyme increases the flow of biologics through tissue under the skin, allowing for rapid dispersal and absorption into the blood stream. And Halozyme says its synthetic enzyme works only in immediate areas where injected with only temporary effects. Pre-filled high-volume autoinjectorThe company licenses it Enhanze technology to pharmaceutical companies for their products. In Sept. 2017, Science & Enterprise reported on Halozyme Therapeutics licensing its Enhanze platform to drug makers Roche and Bristol-Myers Squibb in deals totaling more than $2 billion. The clinical trial enrolled 23 healthy volunteers to test delivery of a sample biologic drug, in this case an immunoglobulin that makes up antibodies in a 10 percent concentration. The active immunoglobulin was delivered with the Enhanze synthetic enzyme using a pre-filled high-volume autoinjector or HVAI device something like injector pens for administering epinephrine to people experiencing severe allergic reactions. The device in this trial delivered 10 milliliters of immunoglobulin in a single dose. Halozyme says the HVAI devices delivered the 10 milliliters of immunoglobulin in 30 seconds. Mike LaBarre, chief technical officer of Halozyme, says in a company statement this speed and volume far exceeds conventional subcutaneous injection methods. “Traditional subcutaneous auto-injector delivery methods,” says LaBarre, “are typically limited to volumes less than 2 milliliters or require long delivery times at slow rates for higher volumes.” LaBarre is the lead presenter of Halozyme’s conference paper tomorrow. In addition, says the company, most skin reactions such as swelling or redness, were mild and resolved within 90 minutes. Nearly all participants (90%) reported no or mild pain after the injections, and all but one of the participants said they would be willing to accept another injection. “Our Enhanze drug delivery and HVAI technologies,” adds Halozyme CEO Helen Torley, “have the potential to rapidly deliver large volume therapeutics subcutaneously with the potential for meaningful clinical benefits.” More from Science & Enterprise:
We designed Science & Enterprise for busy readers including investors, researchers, entrepreneurs, and students. Except for a narrow cookies and privacy strip for first-time visitors, we have no pop-ups blocking the entire page, nor distracting animated GIF graphics. If you want to subscribe for daily email alerts, you can do that here, or find the link in the upper left-hand corner of the desktop page. The site is free, with no paywall. But, of course, donations are gratefully accepted. * * *
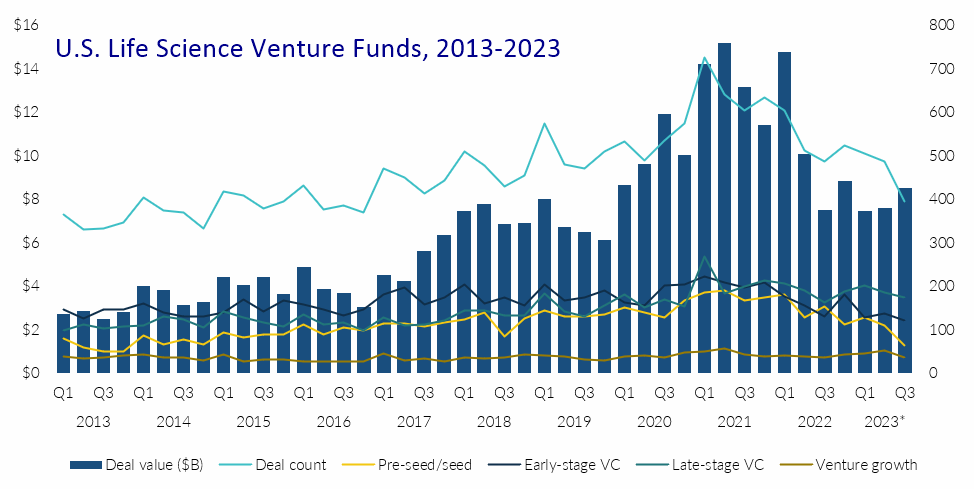 Click on image for full-size view (PitchBook/National Venture Capital Association) 14 Oct. 2023. Venture investments in U.S. life science and biotechnology start-ups in the third quarter of 2023 rose from the previous two quarters to reach pre-pandemic levels. The number of venture deals in this sector, however, continues to slide, with funding for the newest U.S. businesses receiving the lowest percentage of total funds in a decade. The quarterly Venture Monitor report, released this week by venture investment research company PitchBook and National Venture Capital Association, shows total venture investments in U.S. life science and biotechnology start-ups increasing to $8.5 billion for July through September 2023. That quarterly total is the largest so far this year, exceeding the $7.4 billion and $7.6 billion recorded in the first and second quarters of 2023. The $8.5 billion in Q3 is also comparable to the $8.6 billion invested in U.S. life science start-ups in the first quarter of 2020, before the Covid-19 pandemic took hold and venture funding spiked into 2022. The increase in total quarterly investments in this sector, however, masks weaknesses in other indicators. The number of venture deals in the third quarter of 2023 dropped to 395, the fewest transactions per quarter recorded since Q4 2016. Moreover, nearly half (44%) of all life science and biotech investment dollars in the third quarter went to late-stage venture rounds: companies’ third or fourth rounds, called series C and D, with that percentage rising steadily for the past few years. At the same time, the newest U.S. life science and biotech start-ups, those seeking pre-seed or seed funding, experienced continuing declines, completing 63 deals and attracting only 16 percent of all investments, the lowest quarterly percentage since Q3 and Q4 2013. At their current pace, venture investments in U.S. life science and biotech entrepreneurs are expected to reach $31.3 billion this year, the largest annual total since 2019. Total venture transactions for the year at the current pace would reach 1,841, roughly comparable to the 1,868 deals recorded in 2018. More from Science & Enterprise:
We designed Science & Enterprise for busy readers including investors, researchers, entrepreneurs, and students. Except for a narrow cookies and privacy strip for first-time visitors, we have no pop-ups blocking the entire page, nor distracting animated GIF graphics. If you want to subscribe for daily email alerts, you can do that here, or find the link in the upper left-hand corner of the desktop page. The site is free, with no paywall. But, of course, donations are gratefully accepted. * * *
– Sponsored content –  (ulleo, Needpix) 13 Oct. 2023. In 2009, Dysport (abobotulinumtoxinA) was introduced to the US market as an alternative to Botox. Both Dysport and Botox are botulinum toxin-based treatments, primarily indicated for wrinkle reduction and specific movement disorders such as spasticity and cervical dystonia. However, they are derived through unique, proprietary manufacturing processes. Dysport and Botox have different therapeutic effects, including how long they last, how they spread, their side effects, and strength. In this article, we’ll take a look at these differences. What is Botox?Botox (onabotulinumtoxinA) is predominantly recognized as a cosmetic treatment for wrinkles, especially glabellar lines. Beyond its aesthetic applications, Botox functions as a neurotoxin, disrupting chemical communication in cholinergic neurons and affecting signal transmission between these nerve cells and their targets. This action underpins Botox’s broader therapeutic utility. For example, Botox is employed in managing several neuromuscular disorders, including cervical dystonia (involuntary muscle contractions in the neck ) and blepharospasm (eyelid twitch). Moreover, it has shown promise in treating pain-related conditions such as chronic migraines and urological disorders like overactive bladder. Botox offers significant therapeutic advantages to patients across a diverse range of neurological conditions. Dysport and Botox: Both Derived from Botulinum ToxinIn 2009, Dysport (abobotulinumtoxinA) was launched in the US as a counterpart to Botox. Both these treatments have their foundation in botulinum toxin and are primarily prescribed for wrinkle reduction, as well as specific movement disorders including spasticity and cervical dystonia. Yet, they originate from distinct, proprietary production methods. Dysport and Botox have different therapeutic effects, including how long they last, how they spread, their side effects, and strength. Botox (onabotulinumtoxinA), while widely known for its cosmetic use in addressing wrinkles, particularly glabellar lines (frown lines), has more expansive therapeutic implications. It acts as a neurotoxin, interrupting chemical signaling in cholinergic neurons, thus influencing signal relay between these neurons and their respective targets. Such activity forms the basis for Botox’s extended therapeutic applications. Additionally, its potential in managing pain-related conditions like chronic migraines and urological concerns such as an overactive bladder has been recognized. In summary, Botox presents a wide spectrum of therapeutic benefits for numerous neurological conditions. Comparative Dosing between Botox and DysportThe body of clinical research underscores a significant point: Botox and Dysport are not bioequivalent. Specifically, a unit of Botox doesn’t equate to a unit of Dysport in terms of bioactivity. Numerous studies have sought to understand the clinical equivalence of these two formulations for different applications. Generally, the Dysport-Botox ratios employed in these studies have ranged from 3:1 to 6:1, yielding comparably effective outcomes for patients. However, the exact dose equivalency between these botulinum toxin preparations remains an active topic of research and debate. One research focusing on the doses applied for cervical dystonia and blepharospasm found that the Dysport to Botox ratios in clinical practice varied from a minimum of 2:1 to a maximum of 11:1. Notably, 31% of participants were dosed at a Dysport-to-Botox ratio of 5:1 to just under 6:1. Meanwhile, 30% were dosed at a 4:1 to just under 5:1 ratio, and 21% had a dose ratio from 3:1 to just under 4:1. This data suggests that the medical community has yet to converge on a universally accepted conversion ratio that yields consistent results, implying the absence of a straightforward conversion factor. Dysport’s DiffusibilityThere are also observable differences in the physical properties of these neurotoxins. Dysport, as many practitioners have observed, has a propensity to diffuse or spread more readily than Botox. Studies, such as the one conducted by Ranoux et al., highlighted an increased side effect profile with Dysport. The authors hypothesized that this might be tied to Dysport’s higher diffusion rate compared to Botox. This was supported by findings from Nü?gens and Roggenkämper, where Dysport showcased an elevated adverse event profile, particularly a notable rise in ptosis incidents. Further analysis by de Almeida and de Boulle, after reviewing multiple studies on the diffusion characteristics of these neurotoxins, reinforced that Botox tends to diffuse less than Dysport. The diffusibility factor has both benefits and drawbacks, contingent on the application. For instance, the greater diffusion rate of Dysport might be favored for treatments over larger areas, such as in male patients or broader forehead treatments, while Botox is often preferred where precision is paramount. ConclusionIn summary, direct comparisons between Botox and Dysport in clinical trials reveal marked differences in their composition. These disparities manifest in several ways, including in diffusibility, potency, side effect profiles, and duration of effect. It is essential for aesthetic practitioners to remain aware of the non-bioequivalency of these formulations and to exercise caution against direct dose conversions. * * *
 Scanning electron microscope image of a breast cancer cell. (Bruce Wetzel and Harry Schaefer, National Cancer Institute, NIH, Flickr https://flic.kr/p/K4F3ZT) 13 Oct. 2023. Experiences from a rural hospital in North Carolina show hereditary genetic testing of all breast cancer patients is feasible and can result in changes in care for many patients. Researchers from The Outer Banks Hospital in Nags Head, North Carolina and Invitae, a developer of genetic tests and research tools in San Francisco, describe their findings in the 9 Oct. 2023 issue of the journal Annals of Surgical Oncology. Genetic testing is one of the tools for tracking the progression of cancer in patients, particularly when the cancer mutates during the course of treatment. For breast cancer patients, however, guidelines for germline or inherited gene testing are murky. The authors describe conflicts between current guidelines for physicians: The National Comprehensive Cancer Network recommends inherited genetic testing of breast cancer patients with the greatest likelihood of harboring disease-causing genetic variants, such as a family history of the disease, while the American Society of Breast Surgeons recommends inherited genetic testing of all breast cancer patients. For women with breast cancer in rural areas, care is often more difficult to receive. The authors cite data showing less than half of hospitals in upstate New York offer radiation oncology and less than one-third offer breast surgery services. In addition, say the authors, women in rural communities often need to travel further for care, which results in decreased access to radiation therapy and delayed primary therapy. But the authors also note testing for inherited cancer-related genetic changes can provide physicians with better data for decisions on care strategies, such as surgery, radiation, or precision-medicine treatments. Missed positive tests, if using guidelinesThe study team led by Charles Shelton, a radiation oncologist at The Outer Banks Hospital, captured test data and examined medical records from 2019 to 2022, where breast cancer patients at the hospital were eligible for inherited genetic testing as part of their care. Of the 210 eligible patients, 192 or 91 percent, took the test. Participants submitted saliva samples, which in most cases were tested for 47 inherited genes associated with breast and other solid tumor cancers. Participants also received genetic counseling as part of the program. Invitae collected and analyzed the data in the company’s labs, and returned results on average in 14 days. Results show 25 patients or 13 percent tested positive for cancer-related variants, while 63 percent tested negative, and 24 percent had variants of uncertain significance. And if the National Comprehensive Cancer Network guidelines for inherited genetic testing were used, 15 of the 25 patients testing positive would have received the test, while leaving out the other 10 patients. In addition, 129 or two-thirds of those tested used the results to make adjustments in their treatment, from increased surveillance to additional tests, such as MRIs, to decisions to proceed or suspend further treatments or surgeries. “Our study provides a blueprint,” says Shelton in an Invitae statement, “for implementing universal hereditary cancer genetic testing in rural populations who typically see disproportionate care due to access to services and treatment.” Shelton adds that the program of inherited genetic testing “did not lead to under- or overuse of radiation therapy or bilateral mastectomy, thus demonstrating that universal testing enables optimization of clinical care and reduces unnecessary health care resource utilization.” More from Science & Enterprise:
We designed Science & Enterprise for busy readers including investors, researchers, entrepreneurs, and students. Except for a narrow cookies and privacy strip for first-time visitors, we have no pop-ups blocking the entire page, nor distracting animated GIF graphics. If you want to subscribe for daily email alerts, you can do that here, or find the link in the upper left-hand corner of the desktop page. The site is free, with no paywall. But, of course, donations are gratefully accepted. * * *
|
||||
|
Copyright © 2024 Technology News and Literature - All Rights Reserved Powered by WordPress & Atahualpa |
|||||
 RSS - Posts
RSS - Posts
You must be logged in to post a comment.